
My new book, Master Type 1 Diabetes: The Simple, Low-Cost Method to Normalize Blood Sugars, is available in the U.S. on Amazon and internationally on your countries’ Amazon in both Kindle and Print versions. The book incorporates all the new strategies I have learned since my previous book that allowed me to achieve truly normal blood sugars. It also describes why blood sugars can be so difficult to regulate with T1D without these strategies. The ‘Look Inside’ feature on Amazon will allow you to read the first two chapters of the book which gives a complete overview of the book contents.
The Diabetes Control and Complications Trial
It is estimated that iatrogenic hypoglycemia is responsible for 6-10% of deaths in persons with type 1 diabetes (T1D). The Diabetes Control and Complications Trial (DCCT) completed in 1993 showed that “Intensive (insulin) therapy effectively delays the onset of and slows the progression of diabetic retinopathy, nephropathy, and neuropathy in patients with insulin-dependent diabetes mellitus (IDDM).” Consuming a diet high in carbohydrates as was the practice in 1993 (and for most part in 2015) combined with high doses of insulin leads to both hyperglycemia and hypoglycemia. At the beginning of the DCCT, mean insulin doses were 0.62-0.72 U/kg/day. The insulin doses were increased (but not reported) in the intensive insulin therapy group and this increase resulted in both weight gain (with a 33% increase in the mean adjusted risk of becoming overweight) and a three-fold increase in severe hypoglycemia (“severe” meant requiring assistance including seizure or coma). In addition, the glycemic goal of the intensive insulin therapy group was HbA1c < 6.05%, but less than 5% maintained the goal and the mean HbA1c actually achieved was ≈7% (presumably to avoid hypoglycemia). The DCCT clearly showed that improving glycemic control (lowering HbA1c) reduced the incidence and progression of diabetic complications. Another conclusion still applies today, “we recommend that most patients with IDDM be treated with closely monitored intensive regimens, with the goal of maintaining their glycemic status as close to the normal range as safely possible. Because of the risk of hypoglycemia, intensive insulin therapy should be implemented with caution, especially in patients with repeated severe hypoglycemia.”
Asymptomatic Hypoglycemia on The Ketogenic Low Carbohydrate High Fat Diet
In my opinion, the ketogenic low carbohydrate high fat (KLCHF) diet combined with insulin analogs (see blog post #3) developed after DCCT ameliorates the problems of glucose variability and severe hypoglycemia by limiting dietary carbohydrates, thus reducing insulin doses and bringing the mean BG (and HbA1c) toward normal. As I explained in blog post #5 after starting the KLCHF diet, I noticed the symptoms of hypoglycemia suddenly stopped, at least for the first six months. For me, that was marvelous given I had been having them 2-5 times/week. In addition, during the first 3 months on KLCHF, the number of hypoglycemia episodes suddenly decreased. I had one BG reading < 50 mg/dl per week with no symptoms of hypoglycemia. Thus in retrospect, I thought hypoglycemia unawareness was not likely (see below). Getting a reading of < 50 mg/dl on my meter had previously been an extremely rare event. Normally, values that low caused symptoms, so rather than measure the BG, I just treated it. The fact that I could get a reading of < 50 mg/dl with no symptoms was something new and I was seeking an explanation for it. I started reading the literature that I will be presenting below and since then have been leaning toward the idea that my lack of symptoms of hypoglycemia is due to brain utilization of ketones.
Iatrogenic Hypoglycemia
Iatrogenic hypoglycemia is a clinical syndrome of low BG as a result of treating T1D with insulin injections (or pump) typically with symptoms listed below. However, for this discussion I’ll be distinguishing between symptomatic versus asymptomatic hypoglycemia. It is commonly known by clinicians that the glycemic threshold for the development of hypoglycemic symptoms varies depending on the glycemic control of the diabetic patient. The poorly controlled diabetic patient can develop symptoms when the BG falls rapidly from 300 to 100 mg/dl, whereas the tightly controlled patient may not have any symptoms when the BG is 50 mg/dl. This has been described as a shift in the glycemic threshold. The exact mechanism for this phenomenon is not understood. Does the brain adapt to lower BG by increasing glucose transporters? This study found an increase in GLUT3 transporters in rat brain during recurrent hypoglycemia. However in persons with diabetes, some studies have demonstrated increases in steady state brain glucose concentration in type 1 diabetic patients with hypoglycemia unawareness, while others have not.
When hypoglycemia is discussed, often the aforementioned shift in glycemic threshold is set aside and an arbitrary cutoff of 70 mg/dl is used. Thus, hypoglycemia is simply defined as a BG < 70 mg/dl. The symptoms of hypoglycemia are classified as
- Neuroglycopenic – brain dysfunction attributable to central nervous system (CNS) glucose deprivation per se (e.g. confusion, drowsiness, coma, death).
- Neurogenic (or autonomic) symptoms are due to CNS-mediated stimulation of the sympathetic nervous system.
- Adrenergic symptoms (e.g., palpitations, tremor, anxiety) are mediated by catecholamines (epinephrine, norepinephrine) released from sympathetic neurons, the adrenal medullae, or both
- Cholinergic symptoms (e.g., sweating, hunger, numbness) are mediated by acetylcholine released from sympathetic neurons.
Excepting the shift in the glycemic threshold mentioned above, neuroglycopenic and neurogenic symptoms typically occur when BG < 54 mg/dl. Coma can occur when BG < 41– 49 mg/dl. As a reference, a normal BG ≈ 83 mg/dl (4.6 mmol/l). Note the unit conversion: BG in mg/dl ÷ 18 = BG in mmol/l.
In This Blog Post
In this blog post I’ll be reviewing two studies of insulin-induced hypoglycemia in obese subjects undergoing supervised prolonged fasting to assess the ability of the brain to utilize ketones and function appropriately during severe hypoglycemia. Then I’ll review a third study that examined ketone production and oxidation rates as a function of blood ketone levels. I’ll then suggest the possibility that blood ketone levels (specifically beta-hydroxybutyrate (BHB) levels) during nutritional ketosis as a result of consuming a well-formulated KLCHF diet may be adequate to supply the brain with enough energy to prevent symptoms of hypoglycemia. This is just a hypothesis that really needs to be tested in a clinical trial. Finally, I’ll discuss a clinical audit published in 2012 that demonstrated improvement in HbA1c with lower insulin doses by simply lowering dietary carbohydrate intake to 75 grams/day.
Alternate Fuel Utilization by Brain
The first study appeared in a book titled “Cerebral Metabolism and Neural Function” in Chapter 26 titled “Alternate Fuel Utilization by Brain” by George F. Cahill, Jr. and Thomas T. Aoki. The actual study I’m discussing was referenced in the chapter, but I am unable to locate the original study. In brief, three prolonged fasted obese subjects were given 20 units of insulin infused over 24 hours while serum insulin and glucagon, and blood BHB, acetoacetate (AcAc), and glucose levels were monitored. All three subjects developed severe hypoglycemia (mean = 27 mg/dl, lowest was 1 mM or 18 mg/dl), but remained completely asymptomatic. As mentioned above, this degree of hypoglycemia would usually cause seizure, coma, or death. The results are shown in the figure below.
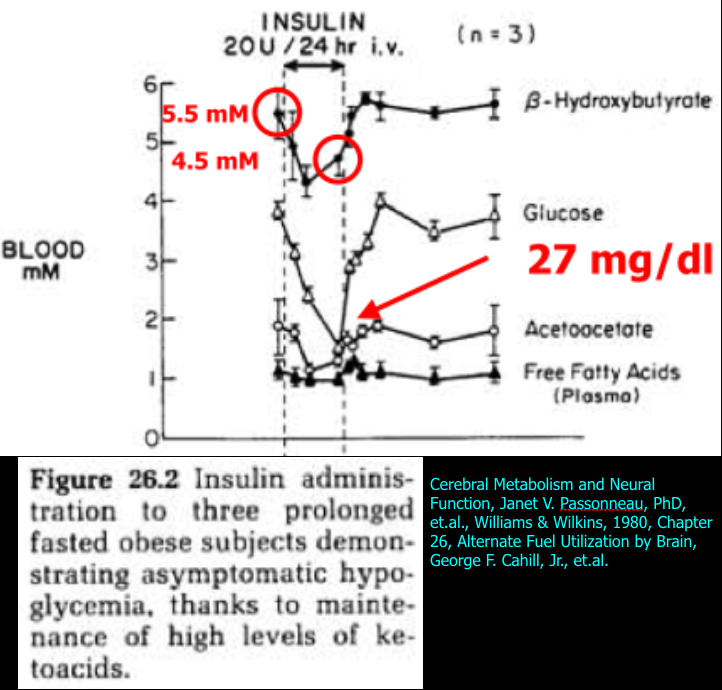
The authors state that the insulin administration inhibited gluconeogenesis (resulting in hypoglycemia), but did not alter BHB or AcAc levels even though they did appear to drop and then rise again during the insulin infusion. Mean BHB concentration started at 5.5 mM, dropped to 4.2 mM, and increased to 4.5 mM at the end of the insulin infusion. Note that in the next study to be discussed, the same fall in BHB and AcAc occurred after insulin administration, but the authors interpreted those results differently by measuring the utilization of ketones by the brain.
Resistance to Symptomatic Insulin Reactions after Fasting
The second study titled “Resistance to Symptomatic Insulin Reactions after Fasting” by Ernest Drenick, et al. was published in The Journal of Clinical Investigation in 1972. This time insulin tolerance tests (0.1-0.2 U/kg single insulin dose) were administered to nine obese men and blood glucose (BG), insulin, and urinary catecholamines were measured both before and after a 2-month fast. Before the fast in response to the insulin tolerance test, the subjects manifested frank hypoglycemic reactions and urinary catecholamines appropriately increased from 61 to 113 ug/24 hr. After the 2-month fast the insulin tolerance tests were repeated, but this time additional tests were performed including blood BHB, and arteriovenous (A-V) differences in BHB and glucose in brain and forearm to measure the utilization of BHB and glucose by brain in comparison with forearm (control). The results are shown in the figure below.
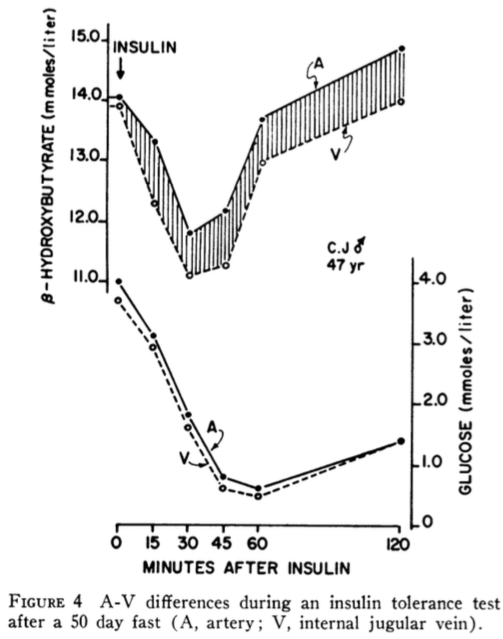
Despite BG concentrations falling to as low as 0.5 mmoles/liter (9 mg/dl), none of the nine subjects exhibited hypoglycemic reactions nor did catecholamines increase in response to hypoglycemia as normally occurs in non-diabetic subjects.
Note that catecholamines (epinephrine and norepinephrine) are usually released by sympathetic nerves and adrenal glands as a normal physiologic response to hypoglycemia to increase hepatic glucose production (via glycogenolysis and gluconeogenesis) to correct the hypoglycemia. The fact that this did not occur in persons without diabetes indicates the brain was not detecting any problems as a result of severely low BG.
I repeat, a BG of 0.5 mmoles/liter (9 mg/dl) would normally cause seizure, coma, death or all three, yet these subjects had no symptoms.
The authors note, “During the post-fast insulin tolerance tests, mean plasma beta-hydroxybutyrate (BHB) decreased from 8.02 to 6.69 mmoles/liter (P <0.01). In another five fasting subjects tested, the A-V difference for BHB across brain increased progressively from 0.21 to 0.70 mmoles/liter whereas across the forearm no consistent uptake could be demonstrated. Simultaneously, the A-V difference across the brain for glucose decreased from 0.24 to 0.07 mmoles/liter of plasma.” The increase in A-V difference for BHB indicates the brain is removing BHB from the arterial blood supply and utilizing it as a fuel. The decrease in A-V difference for glucose indicates the brain is taking up less glucose due to the hypoglycemia. These two observations confirm that BHB becomes the major source of energy for the brain during hypoglycemia after a 2-month fast. The mean plasma BHB decreased from 8.02 to 6.69 mmoles/liter during the post-fast insulin tolerance tests. The authors suggested three possible explanations for this finding. One, inhibition of lipolysis by administered insulin reduces the substrate (fatty acids) for ketone synthesis. Two, insulin directly inhibits the enzyme, 3-hydroxy-3-methylglutaryl (HMG) CoA synthase, for the rate-limiting step in ketone-body synthesis in liver. Finally, tissues (in addition to brain) may need to use ketones as fuel during hypoglycemia, or a combination of these three explanations may explain the decrease in BHB concentration during hypoglycemia. In the discussion section, the authors note, “The findings of this study suggest a possible clinical application. Brittle diabetics, subject to recurrent symptomatic insulin reactions, may possibly benefit from eating ketogenic diets.”
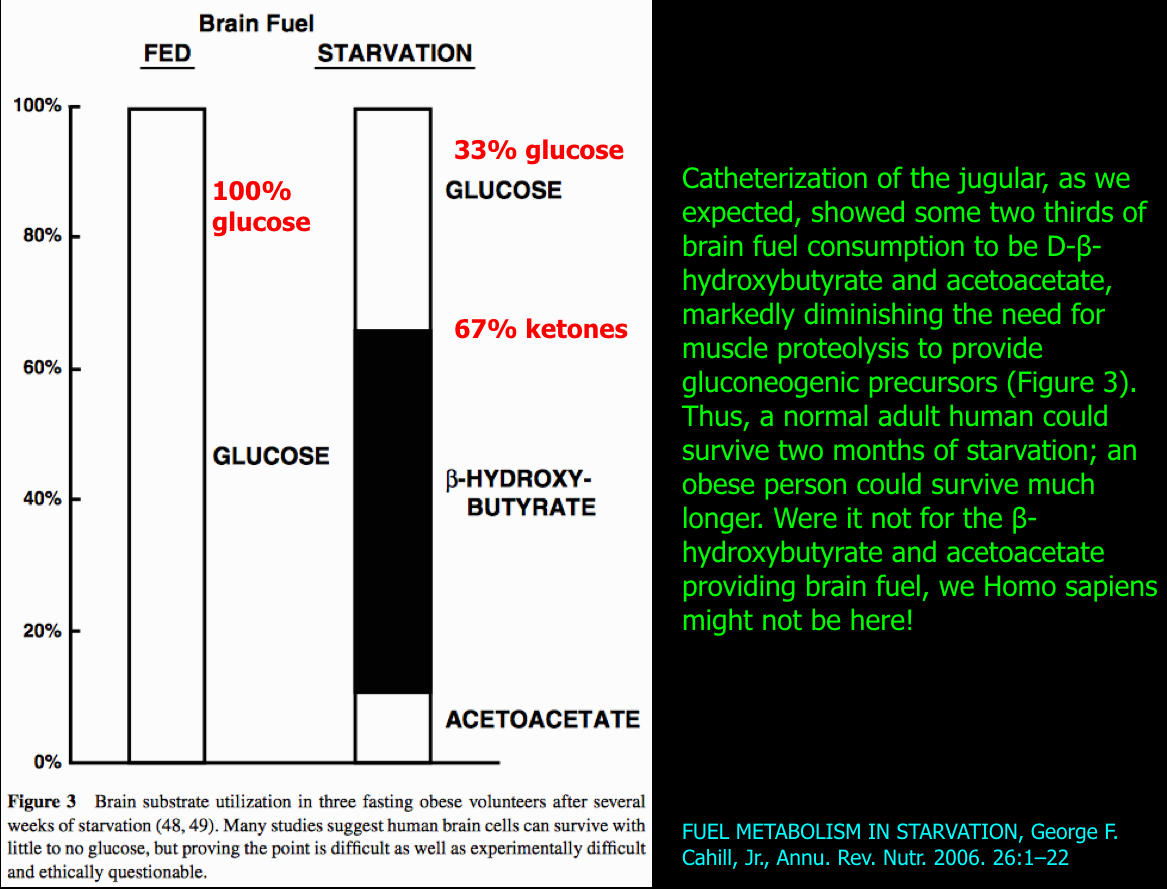
The following figure summarizes the change in brain fuel from 100% glucose on a carbohydrate-containing diet (on left) to that after fasting to 33% glucose and 67% ketones (on right).
Another Look At My Blood Ketone Results
As mentioned in several previous blog posts, the blood BHB typically achieved during nutritional ketosis from the KLCHF diet is 0.5 – 3 mM. In blog post #6, I reviewed my blood BHB levels between Jan. 10, 2013 and August 2, 2014. They ranged from 0.2 – 6.9 mM without (n=10) and with (n=67) coconut oil supplementation. Some were random measurements, but half were measured when my BG was low and having no symptoms of hypoglycemia in order to determine if a correlation existed between blood BHB and BG levels. Of the 77 ketone measurements, 30% were taken when my BG was < 50 mg/dl. Fifty-one percent were taken when my BG was < 70 mg/dl. I had just two hypoglycemic episodes during this 19 month period, but I was not able to measure BHB levels during these two episodes (meter not available). The lowest BG during this time was 26 mg/dl and the BHB level was 3.8 mM, again with no symptoms of hypoglycemia. I repeat, a BG of 26 mg/dl would normally cause seizure, coma, or death, yet I felt fine. In fact, I repeated the measurement thinking the meter strip was bad, essentially the same result 28 mg/dl. Since I was having so few symptoms of hypoglycemia and there was no correlation between the BHB and BG levels, I decided there would be no value in continuing to measure them. Above, I mentioned that insulin directly inhibits ketone-body synthesis in liver. So I plotted my blood BHB data vs the total daily insulin dose on the day of the measurement. The data is shown below.
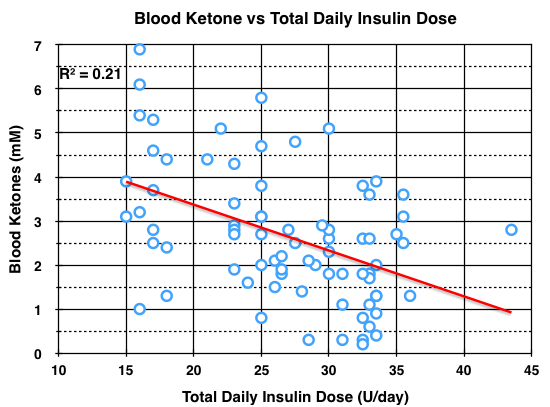
Although there appears to be a wide variation in blood BHB concentration at any given total daily insulin dose, a correlation between blood BHB concentration and total daily insulin dose was statistically significant at the P < 0.05 level. To verify, use R squared = 0.21, R = 0.46, degrees of freedom = 77-2 = 75. Although this may be an intellectual stretch, it is possible that regular exercise which clearly improves insulin sensitivity and reduces insulin doses (in addition to KLCHF), may also improve blood ketone levels and thus improve protection from hypoglycemia.
We saw from the second study (by Ernest Drenick, et al.) above, that BHB levels ranged from 6.69 to 8.02 mM when the subjects were protected from symptoms of hypoglycemia by brain utilization of ketones. So the next question is, are the BHB levels high enough during nutritional ketosis to protect a person with diabetes from the effects of hypoglycemia?
Ketone-Body Production and Oxidation in Fasting Obese Humans
The third study titled “Ketone-Body Production and Oxidation in Fasting Obese Humans,” by G. A. Reichard, Jr., et al. was published in The Journal of Clinical Investigation in 1974. In this study, eight obese volunteers fasted overnight and for periods of 2-24 days. Ketone-body (BHB, AcAc, and total ketones) production and oxidation rates were measured using radioactive isotope labeled BHB and AcAc and plotted as a function of blood ketone concentrations. The arterial blood concentrations of metabolites are shown below (Table II).
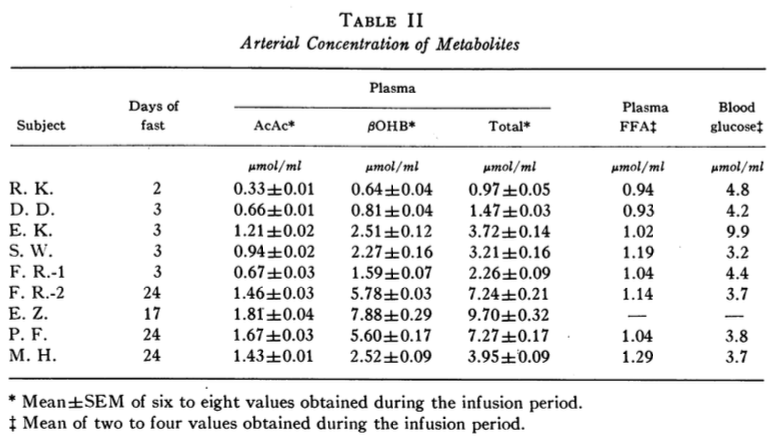
The total ketone-body production and oxidations rates are plotted against the total plasma ketone-body concentrations (Figure 3 below).
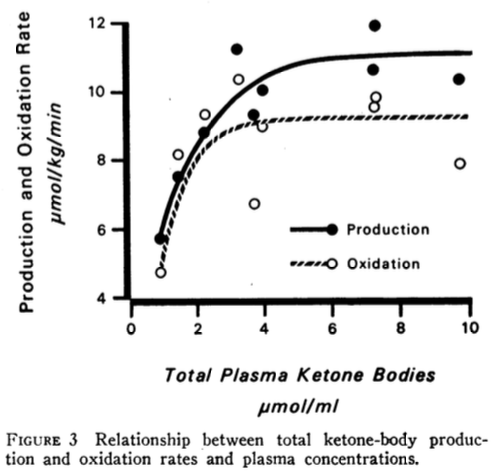
The authors point out that “The maximum rate of total ketone-body oxidation was about 9 μmoles/kg/min and was achieved at a plasma concentration of about 4 μmol/ml (or 4 mM).”
I took the liberty to plot the data shown in Table II above using BHB concentration on the x-axis instead of Total Plasma Ketone Bodies (in Figure 3 above) because blood BHB is what we can measure at home. The ketone-body production and oxidation rates are from Table III in the study (not shown in this blog post). The graph is shown below.
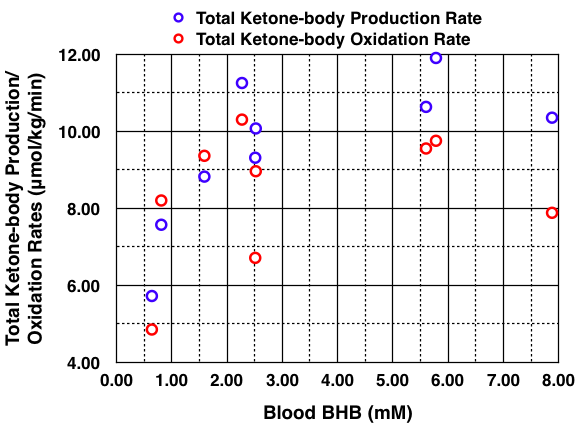
Except for one outlier, the total ketone-body production and oxidation rates reach a maximum at a blood BHB concentration of about 2.5 mM, which is at the upper end of nutritional ketosis (0.5 – 3 mM). The authors state, “This maximum rate of total ketone-body oxidation was achieved in just 2-3 days of fasting. The mean maximum total ketone-body production and oxidation rates were about 11 μmol/kg/min (150 g/24 h) and about 9 μmol/kg/min (129 g/24 h), respectively. Ketonuria accounted for an additional 14 g/24 h (Table IV). The difference between the rate of total ketone-body production and removal is about 7 g/24 h.” Thus, the continued increase in blood ketone concentrations after the first 2-3 days of the fast did not result in any further increase in ketone-body utilization (oxidation), rather the increase in blood ketone concentrations was due to a slightly higher ketone-body production rate above the oxidation rate. “In the subjects fasted for 2-3 days, excluding the diabetic subject E. K., the fraction of the AcAc produced that was immediately oxidized averaged 98.4 ± 4.6% while in those fasted for 17-24 days, the average was 84.2 ± 3.0%.”
Thus, in my opinion, because ketone-body oxidation reached a maximum at a blood BHB concentration of 2.5 mM, it is conceivable that the lack of symptoms I have experienced while in nutritional ketosis could be due to brain utilization of ketones. See blog post #6 for all my blood BHB results. I have not found any published studies measuring brain utilization of ketones while on a KLCHF diet.
The above in no way proves my hypothesis, but on the other hand, I think it should not be completely dismissed either. To be clear, I am striving to avoid hypoglycemia using multiple strategies including adjusting insulin doses, exercise type, intensity, and duration, and relaxing my goal of a normal BG (83 mg/dl) at all times. My current goal is to be satisfied with a BG reading of 70 – 120 mg/dl at all times. Whereas from 2013 through the first half of 2015, I would take a small correction dose of insulin for a BG around 120 mg/dl or higher and this often precipitated hypoglycemia especially if taken after exercise.
Hypoglycemia will continue to occur in persons with IDDM and any additional strategies that might reduce the burden and danger of hypoglycemia would be valuable. Following a KLCHF diet and remaining in continuous nutritional ketosis could be one such strategy. This should be tested in a clinical trial. So far, I have been unable to motivate any research scientists to conduct such a trial.
Is There Another Explanation for Lack of Symptoms of Hypoglycemia?
In this article published in 2005, titled “Mechanisms of Hypoglycemia-Associated Autonomic Failure and Its Component Syndromes in Diabetes” the author, Philip E. Cryer, MD reviews another hypothesis to explain the absence of symptoms of hypoglycemia.
“The concept of hypoglycemia-associated autonomic failure (HAAF) in diabetes posits that recent antecedent iatrogenic hypoglycemia causes both defective glucose counter-regulation (by reducing epinephrine responses to a given level of subsequent hypoglycemia in the setting of absent decrements in insulin and absent increments in glucagon) and hypoglycemia unawareness (by reducing sympathoadrenal and the resulting neurogenic symptom responses to a given level of subsequent hypoglycemia) and thus a vicious cycle of recurrent hypoglycemia. However, the mechanisms of HAAF and its component syndromes are largely unknown. HAAF is now known to be largely reversible, by as little as 2–3 weeks of scrupulous avoidance of hypoglycemia, in most affected patients.”
The following is a more detailed description of the terms used by Dr. Cryer:
- Defective glucose counter-regulation – the normal physiologic response to hypoglycemia is to increase BG by reducing pancreatic beta-cell insulin secretion which in turn increases glucagon secretion by the neighboring pancreatic alpha-cells. A person with IDDM who develops hypoglycemia is unable to alter the action of previously injected insulin and thus the alpha-cells cannot secrete glucagon despite the normal stimulus of hypoglycemia. The other defect present in IDDM is impaired activation of the sympathetic nervous system (both sympathetic nerve activation and epinephrine secretion by the adrenal glands).
- Hypoglycemia unawareness – lacking the symptoms of hypoglycemia and thus being unaware of hypoglycemia which impairs the ability to take corrective behavioral action i.e. to eat food or glucose tablet(s).
- Elevated glycemic threshold – meaning that lower plasma glucose concentrations are required for symptoms to occur and for activation of glucose counterregulatory systems during intensive insulin therapy that effectively lowers overall plasma glucose concentrations.
While antecedent iatrogenic hypoglycemia is the most common risk factor for HAAF, sleep and exercise also increase the risk of HAAF. Awareness of hypoglycemia occurring during sleep is diminished relative to a similar degree of hypoglycemia while awake. Exercise causes increased insulin sensitivity and glucose uptake in muscle for about 16-72 hours after exercise. This can increase the incidence of hypoglycemia if insulin doses are not reduced and/or additional food is consumed. Personally, I avoid any exercise after dinner for this reason. My improved insulin sensitivity lasts about 24 hours which is why I try to exercise every day (to keep insulin sensitivity constant). In addition, I strive to keep my meals and exercise regimen constant from day to day, i.e. diabetes likes consistency.
Addendum dated July 3, 2020 – A study referenced below by Cunnane, SC, et al., published after I wrote this blog post indicates that a blood ketone concentration of 1.5 mM, typical of nutritional ketosis, can only supply enough energy for ≈ 20% of brain metabolism. Thus, hypoglycemia unawareness, rather than brain utilization of ketones, is the most likely explanation for asymptomatic hypoglycemia in those with T1D in nutritional ketosis by following a KLCHF diet. The proper response to asymptomatic hypoglycemia is to avoid hypoglycemia altogether. There are no known benefits of hypoglycemia, but numerous known harms.
Cunnane, SC, et al., 2016. Can Ketones Help Rescue Brain Fuel Supply in Later Life? Implications for Cognitive Health during Aging and the Treatment of Alzheimer’s Disease, Front Mol Neurosci, 9: 53.
This study titled “Hypoglycemia-associated Autonomic Failure in Insulin-dependent Diabetes Mellitus” investigates and reviews HAAF. The study was able to document that one episode of afternoon insulin-induced hypoglycemia lasting less than 2 hours, blunted both the symptoms of and the physiologic responses (an increase in glucagon and epinephrine) to hypoglycemia in persons with diabetes who had no evidence for classic diabetic autonomic neuropathy (a longterm complication of diabetes that can also impair sympathetic responses to hypoglycemia). They conclude that HAAF can result in recurrent severe hypoglycemia thus creating a vicious cycle.
Low Carbohydrate Diet in Type 1 Diabetes
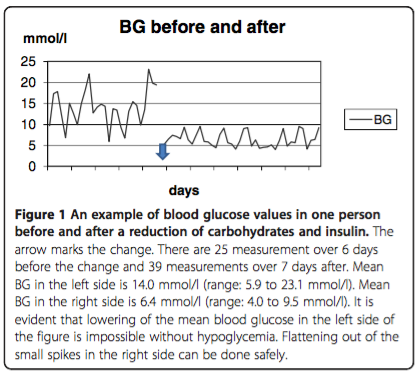
Finally, I will briefly review a report published in 2012 titled, “Low carbohydrate diet in type 1 diabetes, long-term improvement and adherence: A clinical audit.” A total of 48 persons with T1D followed in a diabetes clinic in Sweden who expressed an interest in improving their own glucose control attended a one day course in dietary carbohydrate restriction and insulin dose adjustment with four additional weekly instructional sessions lasting 2-3 hours. Figure 1 below, shows the BG response to reduction in dietary carbohydrate and insulin in one person with T1D.
Adherence was estimated from both HbA1c changes and an individual’s self reported adherence. The HbA1c results over the 4 year followup period for different degrees of adherence are shown in Figure 2 below.
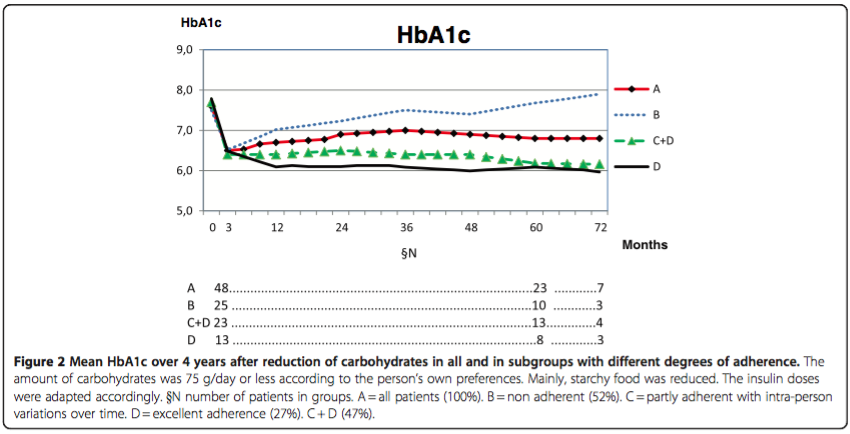
“For individuals with type 1 diabetes one year audit/evaluation of group education in this regimen has shown that the short-time lowering of mean HbA1c by 1 percentage unit and the reduction in mean rate of symptomatic hypoglycemia by 82% was maintained.” I hope it is obvious that adherence to the diet was the major determinate of improvement in glycemic control. Unfortunately, “after 2 years about half of the individuals had ceased adhering.” Additional pertinent comments from the authors: “Only a limited number of patients, about 16-18%, in contact with the present unit (diabetes clinic) have been interested in such a change of diet as described. There is no evidence for the use of the widely recommended high-carbohydrate, low-fat diet in type 1 diabetes. There is no evidence that animal fat in the food should cause cardiovascular disease [13–15]. There is no evidence that protein should cause kidney disease [16]; on the contrary, hyperglycemia gave a 3.5 times higher incidence of albuminuria in DCCT, not protein [1]. There is, however, strong evidence for the aggressive development of damages in all organs in poorly regulated type 1 diabetes [1].”
A measure of the effect of restricting dietary carbohydrates is to compare glycemic control (e.g. HbA1c) and the insulin dose required to achieve it to that on a higher dietary carbohydrate diet. In the DCCT, the base-line HbA1c was 8.8% at an insulin dose of 0.62 U/kg/day in the primary prevention group. The HbA1c decreased to ≈7% with intensive insulin therapy, but the insulin dosage needed to achieve the reduction in HbA1c was not mentioned (presumably more than 0.62 U/kg/day). In the clinical audit from Sweden just reviewed, the base-line HbA1c was 7.6% at an insulin dose of 0.55 U/kg/day. After the first year, the HbA1c decreased to 6.8% with a low carbohydrate diet (75 grams/day) and the insulin dosage decreased to 0.41 U/kg/day despite a reduction in HbA1c.
In Summary
- Iatrogenic hypoglycemia generally limits the ability to achieve and maintain euglycemia in T1D and thus to minimize or avoid microvascular and macrovascular diabetic complications.
- Hypoglycemia results in symptoms at different glycemic thresholds depending on degree of glycemic control. Risk factors for hypoglycemia include recent antecedent hypoglycemia, hypoglycemia unawareness, history of severe hypoglycemia, recent exercise, and sleep.
- HAAF hypothesizes that recent antecedent hypoglycemia causes both defective glucose counter-regulation and hypoglycemia unawareness and thus a vicious cycle of recurrent hypoglycemia. The mechanisms responsible for HAAF are unknown.
- It is estimated that 4-10% of deaths in persons with T1D are due to hypoglycemia and thus it is important to adjust both therapy and glycemic goals to avoid hypoglycemia.
- The DCCT recommendation that most patients with IDDM be treated with closely monitored intensive insulin regimens, with the goal of maintaining their glycemic status as close to the normal range as safely possible is sound.
- In this post, I hypothesized that nutritional ketosis resulting from consuming a well-formulated KLCHF diet may provide adequate blood ketone levels to supply the brain with ketones as an alternate source of energy to provide at least partial protection against the symptoms and adverse effects of hypoglycemia. Just as the HAAF hypothesis will remain a hypothesis until the mechanisms behind it are elucidated, my hypothesis will remain a hypothesis until it can be tested in persons with T1D following the KLCHF diet by measuring brain utilization of ketones during hyperinsulinemic hypoglycemia. I encourage any research scientists in this field to investigate this hypothesis. In light of more recent research by Cunnane, SC, et al. referenced above, asymptomatic hypoglycemia in those in nutritional ketosis by following a KLCHF diet is most likely due to hypoglycemia unawareness than due to brain utilization of ketones. Thus, the safest strategy is to avoid hypoglycemia as much as possible since it has no benefits and many harms.
- The KLCHF diet is a simple and effective lifestyle choice that has been demonstrated to decrease glucose variability, mean BG, HbA1c, insulin doses, and hypoglycemic episodes in the treatment of T1D.

Hi Keith,
I’ve recently come across your blog and have been meaning to post comment to share my experience. I’m 44, T1D and have been trying a ketogenic diet since January. Probably more or less consistently in ketosis for the last 6 months. My overall experience is similar to that of other people who go on the diet – improved glycemic control (HbA1c is finally < 6%) and better overall health. One interesting change is my muscle recovery. I play soccer and previously after a hard game I'd have to pop a couple of ibuprofens in just to sleep through he night. I'd still be in pain the next day. Now, I hard feel any pain at all and 3 hours later I feel like I can play again. Striking!
Anyway, the other two things are T1D related. I'm very interested in the topics you cover your posts, especially in hypoglycemia. Very similar to you, I have cases of what would normally be considered hypoglycemia unawareness. I've been in low 30s and also felt perfectly fine. I've tested my sugar after games where my physical performance was superb and found that I was in the 40s. Similar to you, I just assumed that I was in ketosis and my brain was using ketones as energy. I shared this observation with my endocrinologist (who's generally supportive of my diet), but he would hear none of it and re-iterated the dangers of hypoglycemia (it only takes one episode to die). Unsurprising, I guess. What I find interesting though, is that I still have episodes of hypoglycemia where I'm perfectly aware of it! (Sometimes I would just stop my pump to catch up and sometimes I take a glucose tablet). Anyway, reading your post and your hypothesis, I started wondering if maybe I'm not really that deep in ketosis when I do feel hypoglycemia? It would kind of make sense: if my brain feels that it's running short on energy, it would signal me to get more glucose, but if it's fully energized by ketones, even with a blood glucose of 38, then no need to do anything. Maybe the level of glucose isn't a trigger by itself.
Another issue I'm struggling with is the standard deviation. I'm really struggling in keeping my sugar level between 70 and 90 and while it's nice to think that maybe I'm not in any danger under 50, why test it? One suggestion that my doctor had, which I think is really helpful and I'm trying to make part of my regimen now, is to pump some of the insulin 15 minutes prior to eating. This allows to time its action (I use NovoLog) really well. For example, I would pump 2 units 15 minutes before and then add another 1-2 units with the meal. I've been doing this for a couple of weeks and if done right, the meal doesn't even produce a spike in my glucose chart. I used to chase those post meal highs and was frequently ending up with lows a couple of hours later.
Thanks again for your posts, they are very helpful. This is unchartered territory and it's great to hear about someone else's experience and thoughts!
-Alex
LikeLiked by 1 person
Thanks Alex for sharing your experience with ketosis in treating your T1D, which is the point of this blog. I think what you have accomplished is fantastic! An HbA1c < 6% puts you in a rare group. If my hypothesis is correct, then yes, whether or not we experience symptoms of hypoglycemia will depend on both the level of glucose (degree of hypoglycemia) and ketones. I know from measuring that my blood ketones that I will not always have ketone levels high enough to prevent all symptoms of hypoglycemia. And the brain will always need some glucose, so preventing hypoglycemia is very important. But I think the advantage of nutritional ketosis by following a KLCHF diet is having some protection for those instances when hypoglycemia does happen despite our best efforts to avoid it.
LikeLiked by 2 people
I found your blog yesterday, and I’ve read through all your articles with great interest. I have had similar experiences as you and Alex. My T1D was diagnosed 11 years ago. With carbohydrate restriction I’ve managed to push my hba1c to 5.6 while reducing the amount of hypos. The BG threshold for experiencing hypoglycemia symptoms varies a lot for me too, and I’d always wonder why. Before reading your blog I had not considered ketones as an explanation. Thanks!
LikeLike
Hi Keith
I have a 12 yr old son with T1 diabetes. His last HbA1C was 8.6, previous one was 8.9. A friend has suggested the LCHF diet – I am very keen to do this not only for our son, but myself (and hopefully hubby and daughter as well!) Do you have any advice or opinion on this diet for children? I am a registered nurse and know that this kind of diet is sometimes used in kids with severe epilepsy as a way of reducing seizures. What do you suggest?
LikeLike
In general, the KLCHF diet works for children as well as adults. The ketogenic diet for epilepsy is more restrictive, higher in fat, lower in both protein and carbohydrate, results in higher blood ketone levels (that are needed to reduce/prevent seizures), and has some side effects that are not seen with the less restrictive version for T1D. The diet for epilepsy is followed for months to just a few years generally, because the seizures often do not recur after transitioning off the diet. Dr. Eric Kossoff at Johns Hopkins University is a good resource for those who need more information about treating epilepsy.
For T1D, the KLCHF diet is intended to be followed for life, the carb restriction is about 30 grams/day but can be more for physically active adults, protein is not restricted and ranges from about 1.0 – 1.5 grams protein/kg BW/day. As far as guidance, there are several books available on the Books page of this website that are written for using the KLCHF diet for diabetes. As you will see from both this website and the books, insulin doses need to be reduced and you need to involve your son’s doctor in the process of transitioning to the KCLHF diet. Finally, the TYPEONEGRIT group on Facebook is a support group and information resource for persons with and parents of children with T1D.
LikeLiked by 1 person
Thanks for the quick reply 🙂 Our son uses an insulin pump and we will be exploring ways of getting hold of CGM to help us. My biggest fear is losing him during the night to a severe hypo. It was actually an adult friend of ours with T1D who introduced us to this way of eating so am now on a journey of gathering information to present to J’s endocrinologist and dietician (and praying they will be receptive!) This lifestyle is being taken up by well known Australian elite athletes and the Aussie cricket team doctor is an advocate, so hoping our medical team will be receptive! Again, many thanks 😀
LikeLike
Hi Keith!
I’m a bit late to reply, but here we go anyway 🙂
T1 since >30 years, LCHF/Bernstein/… since Jan 2015.
I have reduced my HbA1c from about 8.0 to 5.5, I have managed to reduce my diabetic retinopathy from early signs to at latest check almost not detectable!!!!
As Alex and you I also find that my hypos are much easier to control, I have had values below 2.7 (48) in which I could not detect any issues. But as Alex states sometimes, I can feel the hypos at much higher values. But still, hypos is always much easier to handle than before low carb. I have assumed that it is ketones that protect me. I do try to eat less than 50 g carb per day but I do not count and I do overeat sometimes. I have never checked ketones.
LikeLike
It is nice to hear others observations of using a ketogenic diet for T!DM. I’m glad you find it both effective and doable.
The reduced symptoms of hypoglycemia on a ketogenic diet I think is related to not only brain ketone utilization, but also due to hypoglycemia unawareness. Now that I understand the term “hypoglycemia unawareness” better from extensive reading on the topic, I think the name is not very descriptive. Essentially, hypoglycemia unawareness occurs when the brain adapts to lower blood glucose (BG) levels and is then able to function properly at lower BG levels. The adaptation can occur rapidly (by the next day, in several human studies ( here and here ). Initially, it was felt to be potentially dangerous because it can lead to more frequent hypoglycemia. However, several animal studies subsequently showed that moderate antecedent hypoglycemia protected the animals from death after severe hypoglycemia. here The role of a ketogenic diet in protecting against hypoglycemia needs to investigated in formal research studies so that medical providers can recommend the diet as standard therapy to their diabetic patients at risk for hypoglycemia.
I checked my ketones for curiosity purposes and the results are shown in blog post #6. here I do not feel it is necessary to check ketones except in two circumstances. If BG is persistently elevated it could be due to diabetic ketoacidosis (DKA) and ketones would be quite high in that situation. Of course, it is better to get the BG under control before reaching the point of DKA, but illness and improper insulin management can cause DKA. Second, if one is not sure whether they are following the diet properly and needs further improvement in glycemic control, measuring ketones and aiming for 0.5 to 3 mM by adjusting dietary carbohydrate and/or protein intake may improve glycemic control.
LikeLike
I cannot tell you how encouraging it is to learn more and more even after 35 out of 38 years of my type 1 diabetic life. Thank you for providing clarity on matters that I was not able to put words to. And, thank you for making sense in a whirl of chaos AT TIMES! KEEP IT COMING PLEASE!
LikeLike
Thanks Molly, I don’t have all the answers, but I’m trying my best to help others.
LikeLike